9月13日,浙江大学微信公众号公布了一项最新科研成果,宣布浙大团队在红斑狼疮研究领域迎来新突破。
文章称,9月10日,由浙江大学良渚实验室、东部战区总医院国家肾脏疾病临床医学研究中心和浙江大学生命科学研究院等单位合作完成一项题为“Loss-of-function mutations in PLD4 lead to systemic lupus erythematosus”的研究论文在Nature发表。该研究首次证实人类PLD4缺陷可导致SLE并阐明了其致病机制,为SLE的精准诊疗提供了重要理论依据。
公开资料显示,系统性红斑狼疮(SLE,简称狼疮)是一种常见的慢性自身免疫疾病,其发病机制复杂,至今尚未完全明确。
文章表示,系统性红斑狼疮不管是在临床症状还是遗传机制,都存在很大的个体化差异。这对认识其发病机制构成了极大挑战。目前,科学界已陆续鉴定30余种由单个基因突变导致的狼疮。研究团队认为,单基因狼疮可以为深入了解SLE的发病机制和开发靶向治疗策略提供重要信息。
通过全外显子组测序,研究人员鉴定到5例狼疮肾炎患者存在PLD4基因突变。PLD4突变属于隐性遗传,患者的两个PLD4等位基因都有突变,而患者父母仅为PLD4突变基因的携带者,没有发病。该项研究首次证实了人类PLD4缺陷与狼疮的关系。
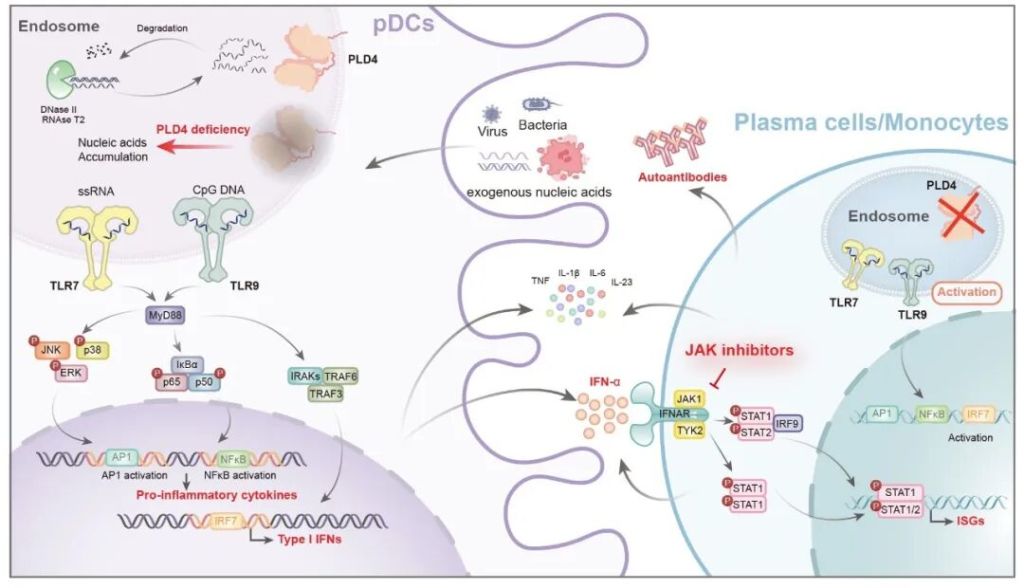
PLD4缺失致使pDCs、B细胞持续活化TLR7/9信号通路,引发自身免疫。浙江大学 图
PLD4在树突状细胞,B细胞和单核细胞中高表达,它们编码一种“单链核酸外切酶”的蛋白。这种蛋白就像一把剪刀,切割降解出现在细胞溶酶体内的单链核酸。研究团队发现:PLD4缺陷患者的PLD4蛋白活性显著降低,就像剪刀“钝”了一样,这一变化介导了过度免疫反应的发生:在这类患者的树突状细胞中,TLR7/TLR9信号通路及其下游I型干扰素的免疫响应明显增强,并伴随多种炎症因子表达水平升高。
为什么仅一个PLD4基因的突变就能引发机体的长期炎症?为了回答这一问题,浙大研究团队从临床转到小鼠实验,摸清从基因突变到临床症状的每一步。也就是PLD4缺失在SLE中的致病机制。
PLD4蛋白下游有两个蛋白TLR7和TLR9,它们参与了细胞中重要的免疫信号通路——I型干扰素信号通路。具体来说,TLR7和TLR9的角色是单链核酸的探测器,如果PLD4功能失活,未被降解的单链核酸就会累积起来,被TLR7和TLR9发现。过多的单链核酸对细胞来说是危险因素,它们有可能是外源的细菌或者病毒带入的,也有可能是从细胞核‘漏’出来的。当TLR7/TLR9感知探测到这些没有被降解的单链核酸,它们会向信号通路下游的分子传递“警报”,引导细胞产生I型干扰素等细胞因子,引发了一系列强烈的免疫反应。
文章介绍,研究还进一步回答了为什么PLD4缺陷的狼疮患者的炎症往往发生于肾脏。这与TLR7和TLR9处于重要的免疫信号通路有关。在动物实验中,Pld4缺陷小鼠表现出典型的狼疮样表型,其中肾脏是PLD4缺陷后受累最为显著的器官,与人类患者的临床肾脏表型高度一致。这些小鼠的肾脏中不仅实质细胞炎症反应增强,还伴有大量免疫细胞的出现,尤其是浆细胞样树突状细胞(pDCs)及浆细胞(PCs)的显著增多。它们进一步招募了更多的免疫细胞,引发了持续的炎症与自身抗体的产生。
研究显示,PLD4缺陷的患者和小鼠,都有I型干扰素信号通路过度激活的现象。这提示可以使用一种已知的免疫抑制剂对这类患者对症治疗。研究团队采用JAK抑制剂巴瑞替尼(baricitinib)对PLD4缺陷小鼠进行干预。在小鼠身上,团队发现它可显著缓解缺陷小鼠体重下降、自身抗体产生及组织炎症等症状。此外,巴瑞替尼还在患者来源的炎症细胞中有效抑制了I型干扰素通路的过度激活,为携带PLD4突变的SLE患者提供了潜在的精准治疗策略。
单基因突变导致的狼疮属于罕见病。对于罕见病的鉴定与治疗,最终是为常见病诊疗提供新的启发。这项研究不仅拓展了对SLE遗传背景的理解,也为未来开展基于基因分型的个体化治疗提供了重要依据。




还没有评论,来说两句吧...